

Publications Abstracts/Posters
Applications and Databases
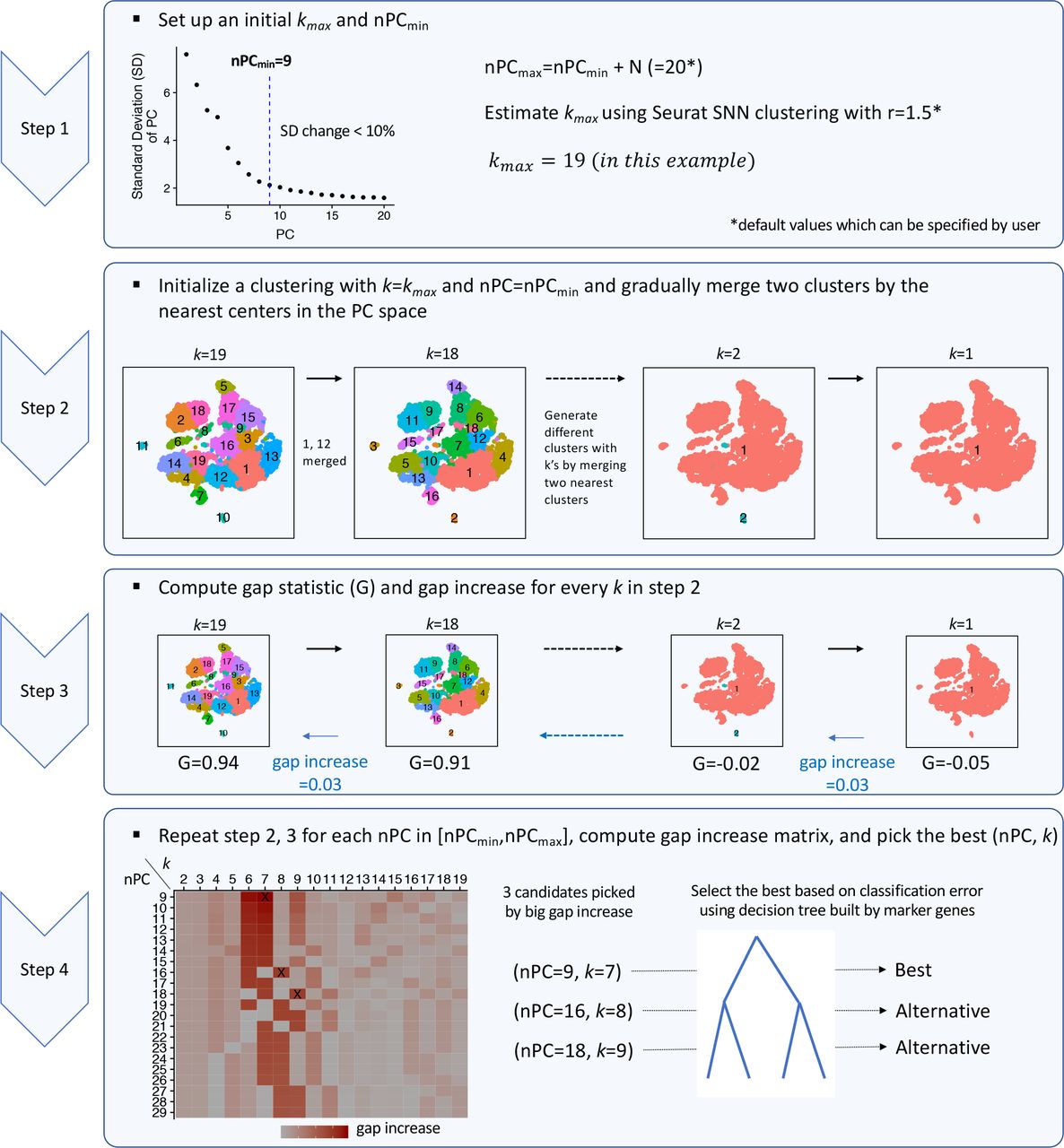
IKAP - Identifying K mAjor cell Population groups in single-cell RNA-seq analysis
|
IKAP - Identifying K mAjor cell Population groups in single-cell RNA-seq analysis. |
 |
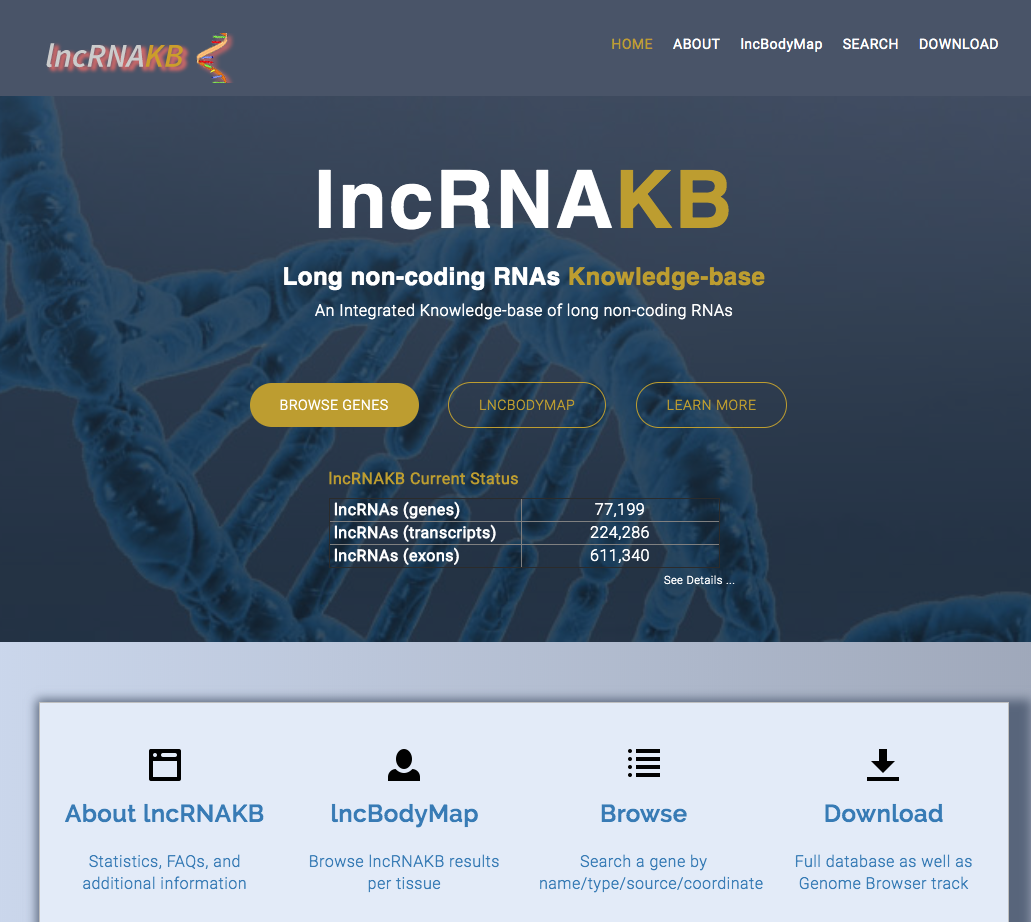
lncRNAKB: An Integrated KnowledgeBase of long non-coding RNAs
|
We assembled the most comprehensive long non-coding RNA knowledgebase (lncRNAKB) by methodically integrating widely used lncRNAs resources. It presents the largest annotation of 77,199 human lncRNAs (224,286 transcripts). We employed Genotype-Tissue Expression (GTEx) project to provide tissue-specific expression profiles and tissue-specificity scores in 31 solid organ human tissues and performed network analysis to identify co-expressed mRNAs that would provide potential understanding on lncRNAs function. It also incorporates coding potential and phylogenetic conservation. Furthermore, using whole genome sequencing data of 652 subjects from GTEx, we calculated expression quantitative trait loci (cis-eQTL) regulated lncRNAs in all tissues. [ lncRNAKB.org ] [ GitHub ] [ OSF ] [ Publication ] |
 |
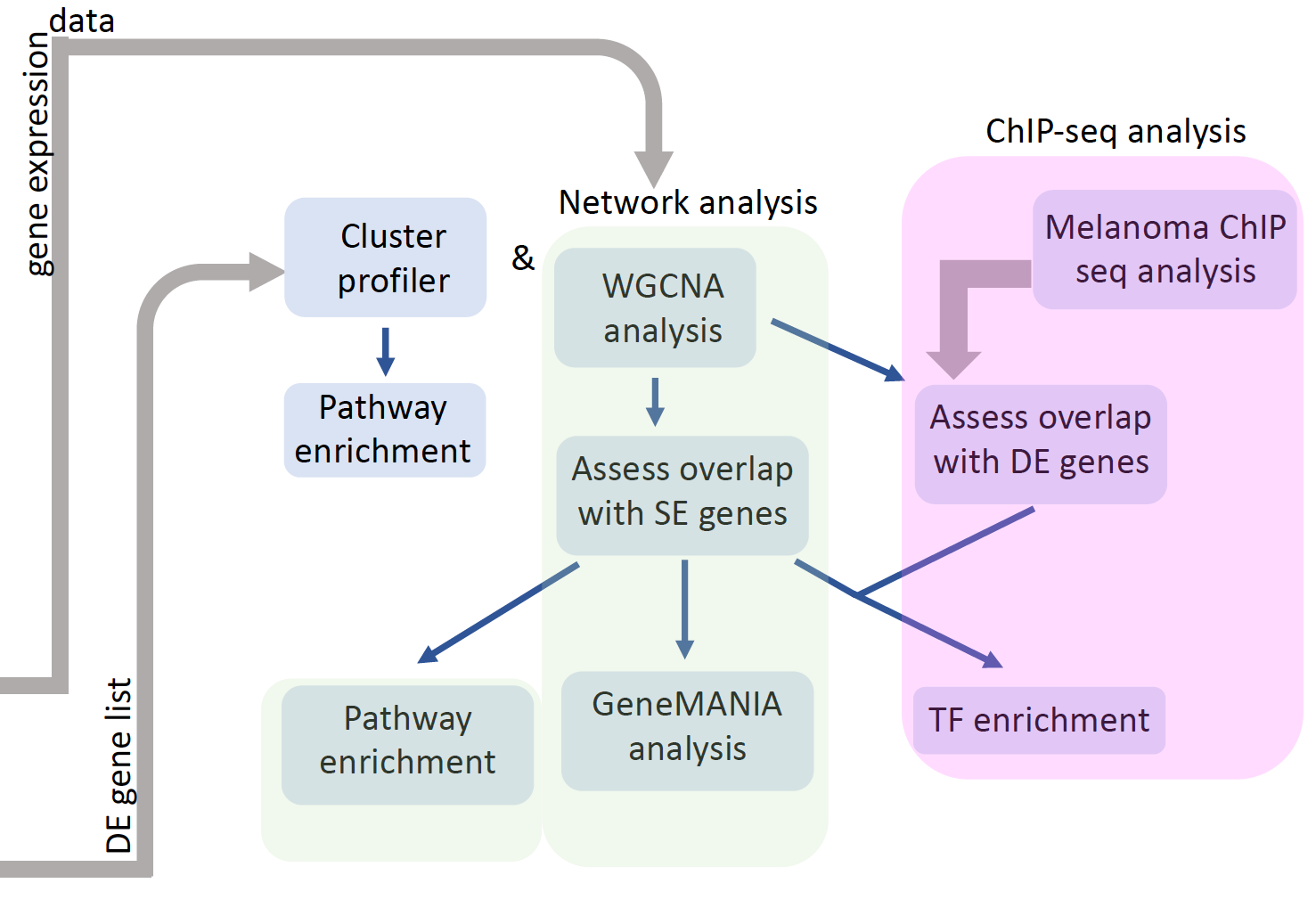
Cancer progression analysis using network biology
|
Using a network biology platform that uses transcriptomic profiles in early and late stages of melanoma, the goal is to identify mechanisms that drives melanoma progression.
We have performed a consensus network analysis of RNA-seq data from clinically re-grouped melanoma samples to identify gene co-expression networks that are conserved in early (stage 1) and late (stage 4/invasive) stage melanoma. Overlaying the fold-change information on co-expression networks revealed several coordinately up or down-regulated subnetworks that may play a critical role in cancer progression.
|
 |
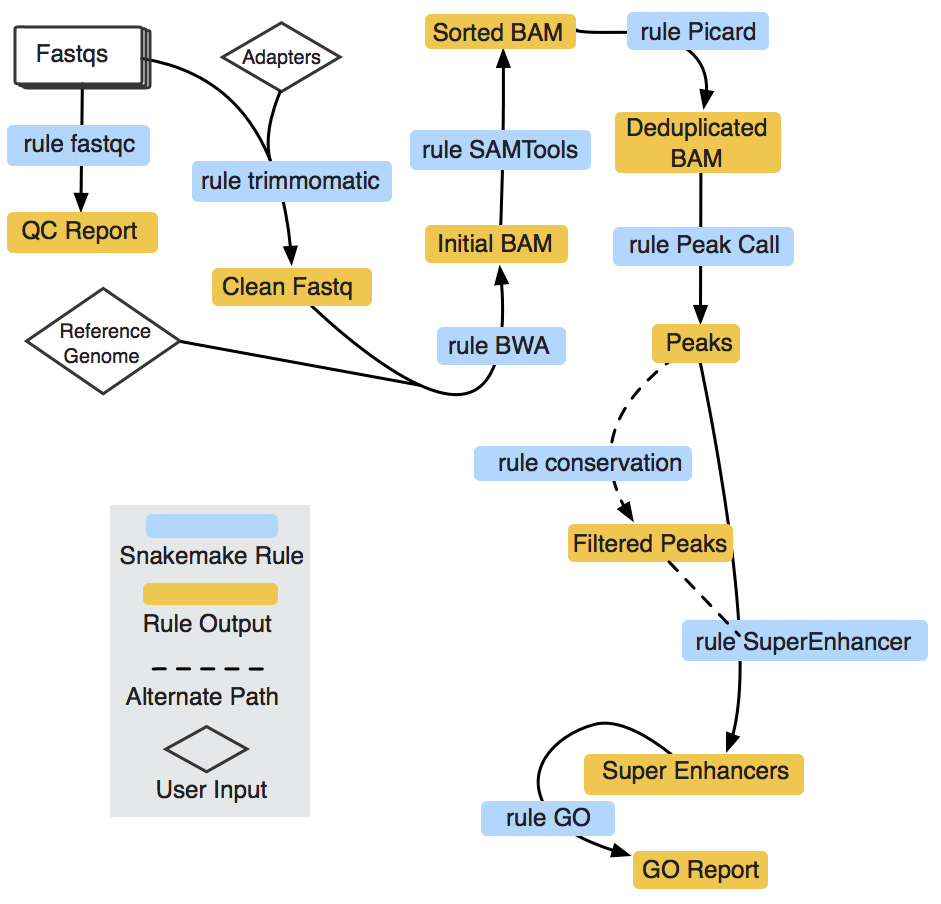
SEpipe: An Automated Cloud-based Super Enhancer Analysis Pipeline.
|
A super enhancer (SE) is a genomic locus on the genome that has a cluster of enhancers in close proximity. Identification of SEs is a three-step process that begins with the identification of enhancers followed by clustering or stitching enhancers that lie is close physical proximity. An accepted parameter for scanning enhancers for clustering or stitching is 12.5 kilo bases (kb). In the final step, the stitched enhancer regions are ranked by signal enrichment to identify putative SE's. A simple visualization plot known as hockey-stick plot is generated, an inflection point in this plot is identified as cut-off to generate a list of putative enhancers. Here we present a cloud-based, fully automated pipeline to identify SEs from H3K27ac based sequencing data sets. In addition to standard SE analysis, we also incorporate the prior knowledge of conservation. Using conservation knowledge, users can reduce false positives in the SE identification process.
|
|
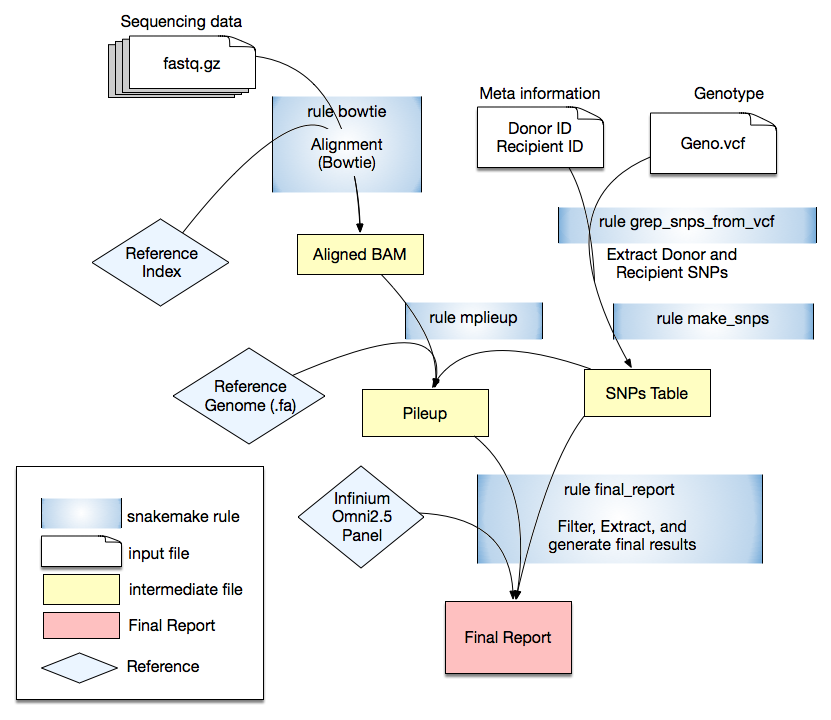
cfCloud: A Cloud-based Workflow for Cell-Free DNA Data Analysis.
|
Cell-free DNA (cfDNA) is double stranded, non-randomly fragmented short (<200bp) DNA molecules circulating in the blood stream as a result of apoptosis, necrosis or active secretion from cells. The amount of cfDNA in blood increases dramatically with cellular injury or necrosis and therefore, can be used as a biomarker as a non-invasive prenatal testing, tumor-derived DNA in plasma, or monitoring the graft health in an organ transplantation. The Genome Transplant Dynamics, a rigorous and highly reproducible universal NGS based method, has been commonly used to utilizes genotype information differences in recipient and donor to quantify donor derived cell-free DNA percent. Here we implement a fully automated Snakemake pipeline on-premise as well as a Cloud implementation to systematize the quantification of donor derived cell-free DNA amount.
|
|
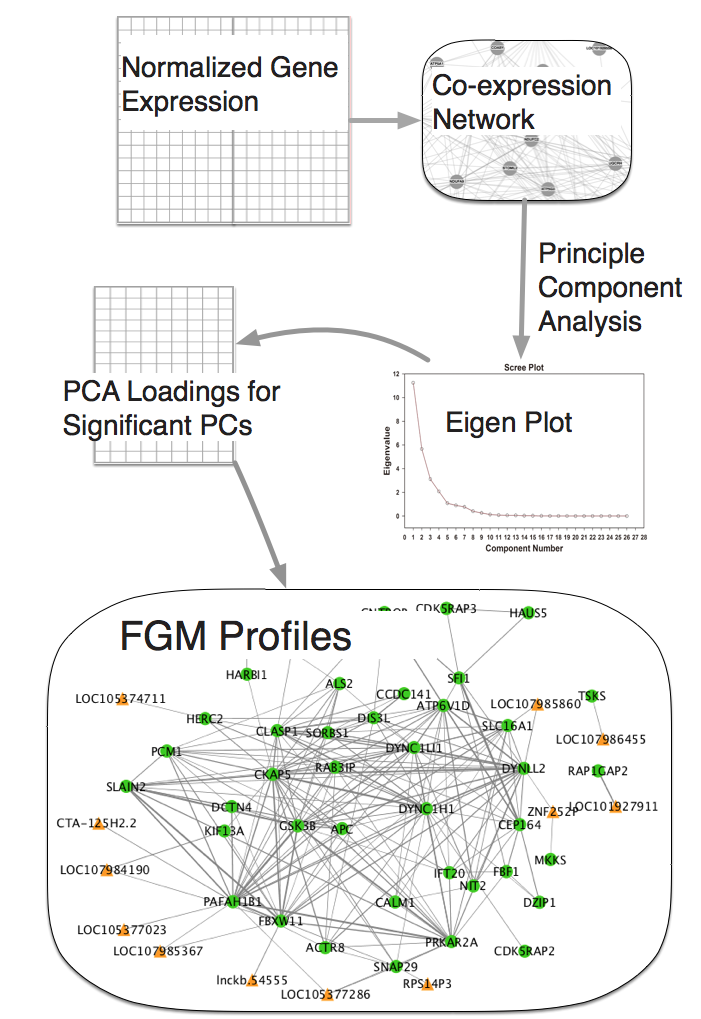
rFGM: Co-regulation Network Inference using an efficient FGM Profiling.
|
rFGM: Co-regulation Network Inference using an efficient FGM Profiling.
Functional genomic mRNA (FGM) profiling [26] is an approach that generates a co-regulation network based on transcriptomic profiles. Given a large number of transcriptome profiles (generally in hundreds) as the primary input, the FGM profiling approach builds a correlation network and then applies principal component analysis (PCA) on the network. Traditionally PCA is applied at sample level to identify outliers or at the gene level to identify cell types in single cell RNA-seq. Application of PCA at gene level in bulk RNA-seq experiments may be driven by highly expressed genes which will overshadow the characterization of genes with low or moderate expression, a majority of this class, for instance, comes from non-coding transcriptomes. FGM profiling sought to overcome this issue. The gene that heavily contributes to the significant principal components (PCs) of the co-expression networks are expected to be enriched for biological pathways. The application of PCA on the co-expression matrix is computationally expensive.
Here we apply approximate but highly accurate PCA computation tools to perform FGM profiling in a reasonable time frame (in few hours).
|
|
Rabbit Genome Study
|
TRANSCRIPTOMICS AND PROTEOGENOMICS STUDY OF RABBIT (ORYCTOLAGUS CUNICULUS) |

|