

Research Projects
The broad research goal of these projects is to simulate a cell membrane on the atomic level. Such computer studies hold extraordinary promise: in applied areas such as drug design, they would provide a detailed picture of drug binding and transport through the membrane; on the fundamental level, they would answer questions concerning cell fusion and signal transduction, and are a valuable complement to experiment.
There are three specific project areas:
1. Understand Model Membranes
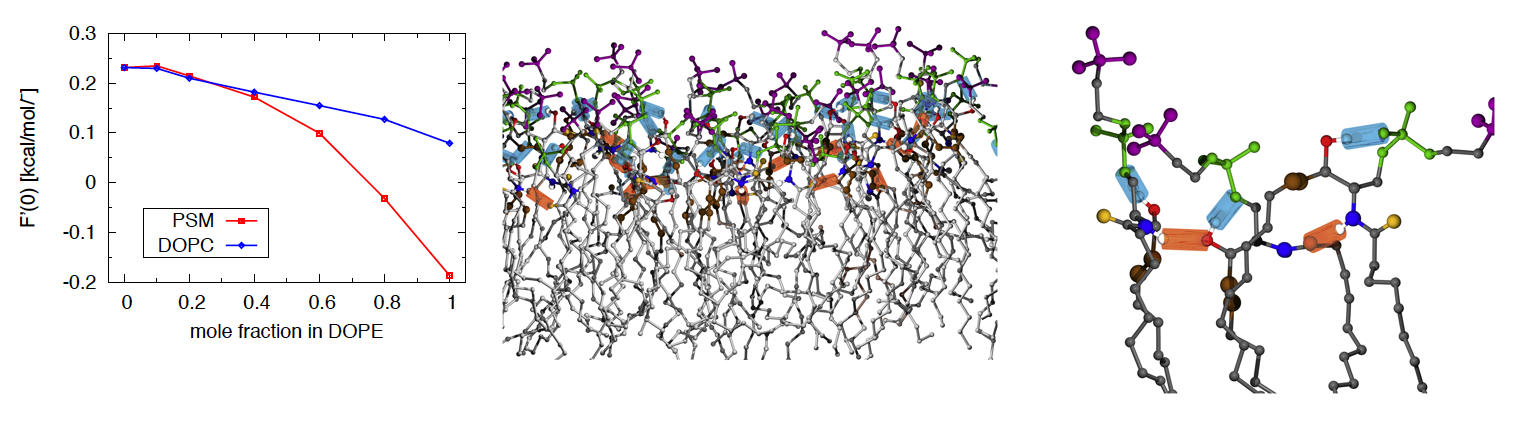
"Nonadditive Compositional Curvature Energetics of Lipid Bilayers," A. J. Sodt, R. M. Venable, E. Lyman and R. W. Pastor, Physical Review Letters, 117 (13) (2016). [DOI]
"CHARMM All-Atom Additive Force Field for Sphingomyelin: Elucidation of Hydrogen Bonding and of Positive Curvature," R. M. Venable, A. J. Sodt, B. Rogaski, H. Rui, E. Hatcher, A. D. MacKerell Jr, R. W. Pastor and J. B. Klauda, Biophysical Journal, 107 pp. 134-145 (2014). [DOI]
Experiments on bilayers and monolayers containing lipids such as dipalmitoylphosphatidylcholine (DPPC) provide the essential target data for this project. These include NMR relaxation times and deuterium order parameters, X-ray structure factors, lateral diffusion constants, elastic constants, and dipole potentials, and phase transitions. Models for experimental observables (e.g., NMR spin lattice relaxation) are also developed both to supplement experiment and to provide further insight into the fundamental physics of membrane structure and dynamics.
2. Develop Simulation Methodology
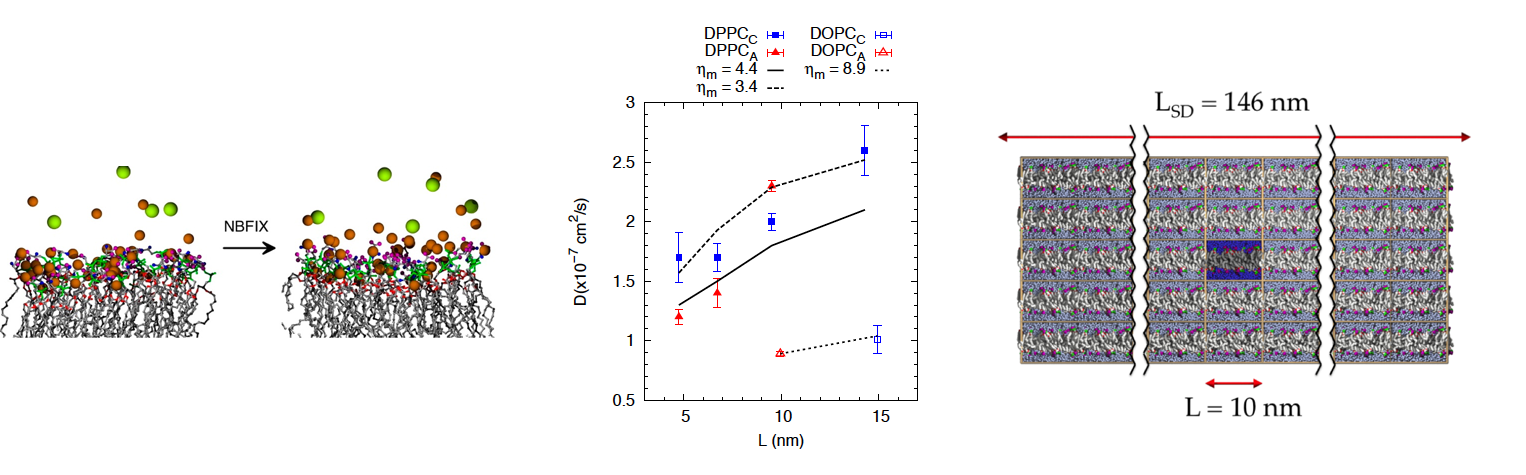
"Simulations of Anionic Lipid Membranes: Development of Interaction-Specific Ion Parameters and Validation using NMR Data," R. M. Venable, Y. Luo, K. Gawrisch, B. Roux and R. W. Pastor, Journal of Physical Chemistry B, 117 pp. 10183-10192 (2013). [DOI]
"Lipid and Peptide Diffusion in Bilayers: The Saffman–Delbrück Model and Periodic Boundary Conditions," R. M. Venable, H. I. Ingólfsson, M. G. Lerner, B. S. Perrin, B. A. Camley, S. J. Marrink, F. L. H. Brown and R. W. Pastor, The Journal of Physical Chemistry B, 0 (0) pp. Article online ASAP (2016). [DOI] [URL]
The current principal focus of this project is the development of molecular mechanic force fields (FF) suitable for atomic level simulations of simple and complex bilayers, including glycolipids. To this end a combination of high level quantum mechanical calculations and molecular dynamics (MD) simulation are carried out on model compounds, compared with experiment, and then applied to lipids and related compounds. Development of the simulation programs such as CHARMM (Chemistry at HARvard Molecular Mechanics) is also carried out as part of this project. Such efforts have included development of algorithms for simulating interfacial systems, calculation of long-range forces, and stochastic dynamics.
3. Simulate Complex Membranes
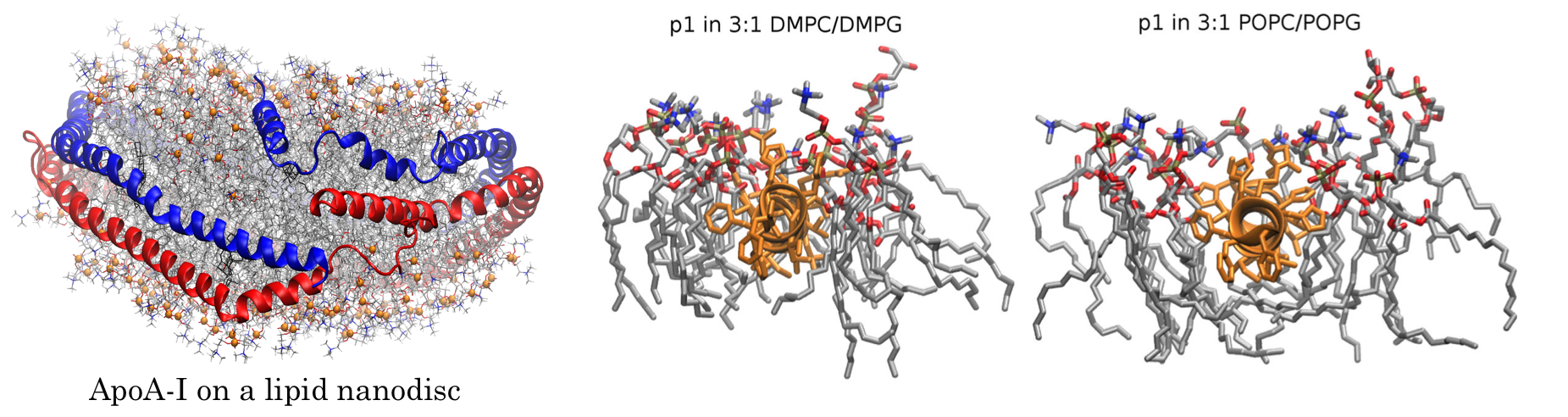
"The Curvature Induction of Surface-Bound Antimicrobial Peptides Piscidin 1 and Piscidin 3 Varies with Lipid Chain Length," B. S. Perrin Jr., A. J. Sodt, M. L. Cotten and R. W. Pastor, Journal Of Membrane Biology, 248 (3, SI) pp. 455-467 (2015). [DOI]
These include bilayers with complex lipids (e.g., gangliosides and phosphoinositides), other components including cholesterol, sugars such as trehalose, viral fusion peptides, and bacterial toxins. This work naturally follows from 1 and 2, and, ultimately, will become the primary focus of the section.